













Qué es el remdesivir, el primer fármaco autorizado contra COVID-19 que Trump quiere acaparar
 Qué es el remdesivir, el primer fármaco autorizado contra COVID-19 que Trump quiere acaparar
Qué es el remdesivir, el primer fármaco autorizado contra COVID-19 que Trump quiere acaparar





Francisco López-Muñoz, Universidad Camilo José Cela and Jose Antonio Guerra Guirao, Universidad Complutense de Madrid
El 1 de mayo, la Agencia para los Alimentos y Medicamentos de Estados Unidos (FDA) emitió una “autorización de uso urgente” para el tratamiento de casos graves de COVID-19 con el antiviral remdesivir. Y el 25 de junio, la Agencia Europea de Medicamentos (EMA) recomendaba a la Comisión Europea su “autorización condicional” para estos pacientes.
El remdesivir ha generado polémica en los últimos días por dos motivos. En primer lugar, su precio de comercialización será de algo más de 2 000 euros por paciente, lo que ha abierto un nuevo debate social, de naturaleza bioética, sobre adquisición de medicamentos y salud pública.
Pero, además, el gobierno de EE. UU. ha adquirido el 90 % de la producción de este fármaco para los próximos tres meses.
¿Qué es el remdesivir y cómo funciona?
El remdesivir es un profármaco (una sustancia inocua que se transforma en fármaco dentro del organismo cuando es metabolizada) antiviral. Pertenece a la familia de los análogos de los nucleótidos y actúa inhibiendo una enzima del virus indispensable para su multiplicación. Ha demostrado tener actividad in vitro contra el SARS-CoV-2:
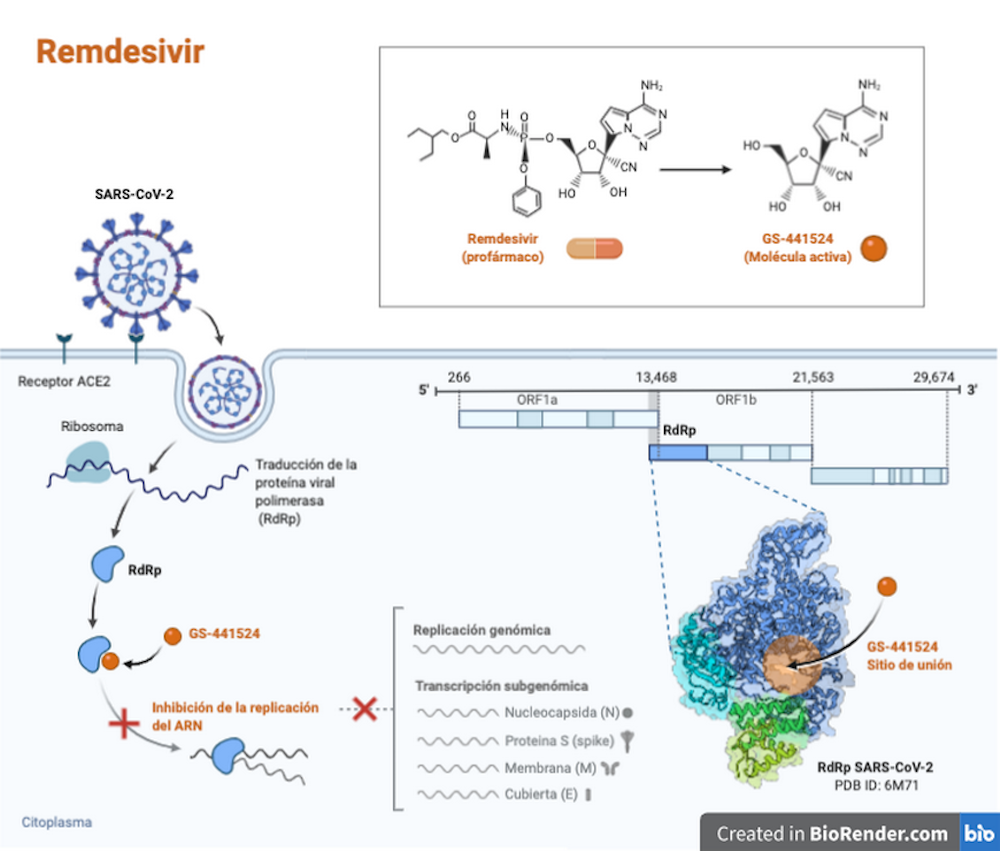
El remdesivir se desarrolló como tratamiento para la infección por el virus del ébola, pero presenta también actividad in vitro frente a otros virus, incluidos algunos coronavirus como el causante del MERS y el SARS-CoV-2.
¿Es eficaz y seguro en pacientes con COVID-19?
A finales de abril de 2020 se publicaron en The Lancet los resultados preliminares de un estudio aleatorizado, doble-ciego y controlado, realizado en Hubei (China). En este estudio se incluyeron 237 pacientes (158 tratados con remdesivir y 79 tratados con placebo) con infección grave por SARS-CoV-2.
El estudio evaluó el “tiempo hasta la mejoría clínica”. Aunque los resultados no fueron estadísticamente significativos, se observó que el “tiempo hasta la mejoría clínica” fue menor en los pacientes que recibieron remdesivir en el marco de los primeros 10 días desde el inicio de los síntomas.
Sin embargo, otros estudios, como el NIAID-ACTT-1 –otro ensayo clínico multinacional, aleatorizado, doble-ciego y controlado–, en el que se evaluó la eficacia y seguridad del remdesivir en pacientes hospitalizados con neumonía por SARS-CoV-2, sí obtuvieron resultados estadísticamente significativos.
En este caso, los pacientes que recibieron remdesivir tuvieron un “tiempo hasta la mejoría clínica” un 31 % más corto que los que recibieron placebo. Concretamente, “el tiempo medio hasta la mejoría clínica” fue de 11 días para los pacientes tratados con remdesivir, frente a los 15 días en el grupo de pacientes que recibieron placebo.
Los resultados también sugirieron un beneficio en la supervivencia, con una tasa de mortalidad de 7,1 % en el grupo que recibió remdesivir, frente al 11,9 % en el grupo placebo. Esta mejoría se observó sobre todo en aquellos pacientes con insuficiencia respiratoria (déficit de oxígeno en la sangre), pero que no requerían de respiración mecánica o extracorpórea.
Con respecto a la tolerabilidad, el perfil de seguridad de remdesivir no se ha caracterizado de manera completa. La mayor parte de la experiencia clínica se relaciona con su uso en el control del ébola, que difiere profundamente del SARS-CoV-2. Sin embargo, hasta ahora no hay hallazgos de seguridad que impidan su uso en pacientes con COVID-19.
Entre las preocupaciones a tener en consideración en los pacientes tratados con el remdesivir se encuentran la función renal y hepática, que deben ser monitorizadas antes y durante el tratamiento. Los datos de uso compasivo en 61 pacientes graves de COVID-19 tratados con el remdesivir, publicados en abril en el New England Journal of Medicine, pusieron de manifiesto que el efecto adverso más común fue el aumento de los niveles de las enzimas hepáticas, observado en el 23 % de los pacientes, siendo la hipotensión arterial el segundo (8 % de los pacientes).
Por otro lado, el remdesivir presenta algunas similitudes estructurales y funcionales con otro medicamento antiviral, el tenofovir, que ha confirmado ser nefrotóxico, tanto en pacientes con hepatitis crónica tipo B como en modelos animales. En humanos, el remdesivir se elimina en gran medida a nivel renal, lo que podría ocasionar una acumulación orgánica del medicamento en aquellos pacientes con insuficiencia renal.
También se han informado efectos adversos gastrointestinales de menor intensidad, como náuseas y diarrea, en un 3 % a 5 % de los pacientes tratados, aunque estos efectos también podrían asociarse a la sintomatología de la COVID-19.
¿Cómo y cuándo se administra?
El remdesivir se administra a través de una vía intravenosa por goteo. Su uso se limita a los centros hospitalarios en los que los pacientes pueden ser vigilados estrechamente, monitorizando la función hepática y renal, antes y durante el tratamiento. El tratamiento debe comenzar con una infusión de 200 mg el primer día, seguida de una infusión de 100 mg al día durante, al menos, 4 días más. Nunca durante más de 10 días.
¿Por qué su autorización condicional?
En abril de 2020 se comenzó la evaluación de los datos de calidad, fabricación, preclínicos y clínicos preliminares, así como los datos sobre seguridad procedentes de la experiencia adquirida con el uso de este medicamento en los programas de uso compasivo. Con todos estos datos, la EMA ha propuesto la autorización condicional de este medicamento, lo que implica que se tienen que ampliar los datos de eficacia y seguridad, por lo que hay que continuar investigando con este fármaco.
La autorización condicional de un medicamento significa que se considera que satisface una necesidad médica no cubierta en la medida en que el beneficio para la salud pública de su inmediata disponibilidad es superior a la incertidumbre derivada de la limitación de los datos disponibles.
Sin embargo, el laboratorio comercializador (en este caso Gilead Sciences), debe comprometerse a proporcionar más datos clínicos que completen la información actual sobre eficacia y seguridad del medicamento. En este sentido, solo cuando estos datos sean positivos se concederá la autorización completa y definitiva. En caso contrario, se retirará del mercado.
¿Qué podemos esperar de este fármaco antiviral?
El remdesivir ha abierto una importante puerta al tratamiento de la COVID-19. Es el primer agente autorizado que actúa directamente sobre el virus. Pero, aunque se ha demostrado inicialmente su eficacia en pacientes graves o muy graves, su perfil de seguridad no está totalmente esclarecido.
En la actualidad, hay más de siete ensayos clínicos en el mundo que están evaluando este antiviral, y la Universidad Johns Hopkins considera al remdesivir como el fármaco más prometedor en estos momentos para el tratamiento de la COVID-19.
Por todos estos motivos, la comunidad científica, junto con las autoridades sanitarias y regulatorias, deben vigilar estrechamente todos los datos clínicos que se vayan aportando en los próximos meses para confirmar definitivamente la idoneidad de este tratamiento.
Francisco López-Muñoz, Profesor Titular de Farmacología y Vicerrector de Investigación y Ciencia, Universidad Camilo José Cela and Jose Antonio Guerra Guirao, Profesor de Farmacología y Toxicología. Facultad de Farmacia. Universidad Complutense de Madrid., Universidad Complutense de Madrid
This article is republished from The Conversation under a Creative Commons license. Read the original article.


 no avanza es porque algunos no quieren tocar las AFP\"")




