












Por qué sufro la enfermedad más bonita del mundo (y la más cara de tratar)
 Por qué sufro la enfermedad más bonita del mundo (y la más cara de tratar)
Por qué sufro la enfermedad más bonita del mundo (y la más cara de tratar)



T13 En Vivo
Me llamo Antonio y padezco atrofia muscular espinal (AME, para los amigos). Mi esperanza de vida era de pocos años cuando nací, hace ya casi medio siglo. Y todo porque "me tocó" una enfermedad rara, neurodegenerativa, que está entre las enfermedades genéticas con mayor mortalidad asociada.
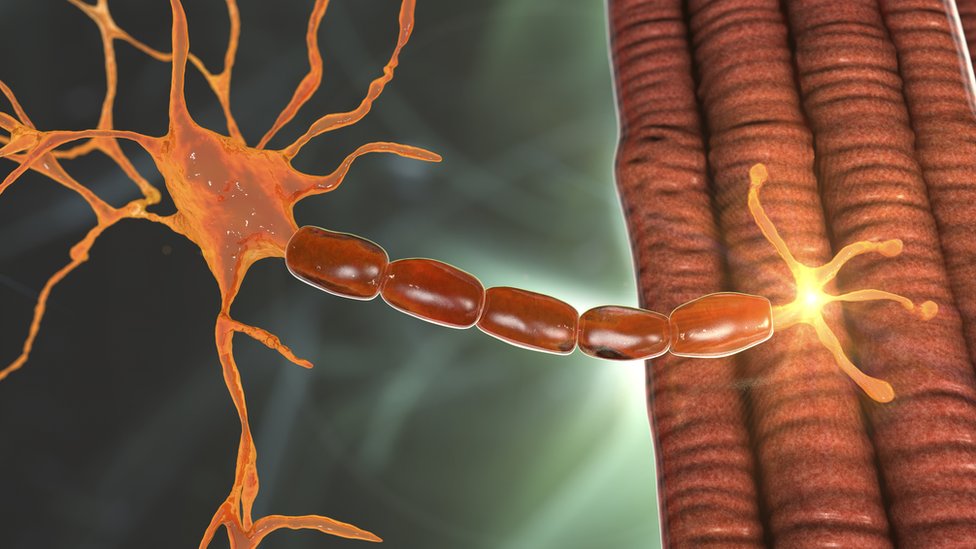
Lo que ocurre, explicado de una manera sencilla, es que mis motoneuronas (los "cables" que unen la médula espinal con los músculos) se van degenerando poco a poco y acaban muriendo.
Esto me ha tenido sentado en una silla de ruedas la mayor parte de mi vida. Y con el tiempo, la afectación de los músculos respiratorios puede hacer que llegue el fatal desenlace.
Pero como además soy científico, a todo lo anterior debo añadir que he descubierto que la AME es la enfermedad más bonita del mundo… a nivel molecular.
¿Cómo se produce la atrofia muscular espinal?
La AME es una enfermedad genética producida por la mutación o pérdida de un gen llamado SMN1.
Si nuestra madre y nuestro padre tienen una copia mutada (una de las dos copias de las que dispone cada uno), y nosotros tenemos la mala suerte de heredar estas dos copias defectuosas, lo más probable es que padezcamos AME.
Este gen contiene la información para guiar en la construcción de una proteína llamada SMN. Su nombre proviene de las sugerentes siglas del inglés survival of motor neuron (supervivencia de la motoneurona), y desempeña funciones muy importantes en nuestro organismo.
De hecho, sin SMN no sobreviven las motoneuronas, ni tampoco hay posibilidad de vida en sí misma.
Se debe a que estas células son esenciales para llevar a cabo movimientos musculares, incluidos los del diafragma o los de la faringe, que nos permiten respirar y comer.
Pero entonces ¿cómo sobrevivimos los pacientes de AME? Pues porque resulta que en el genoma humano hay un segundo gen, llamado SMN2, que también permite construir esa misma proteína SMN. SMN1 y SMN2 son casi idénticos, pero SMN2 se distingue en cinco de las más de 40 000 letras o nucleótidos de las que se componen estos genes.
La existencia de genes prácticamente idénticos es algo muy raro en el genoma humano. De hecho, SMN2 ni siquiera existe en otros animales: es único de nuestra especie.
Un salvavidas
Esas mínimas diferencias entre los dos genes hacen que la proteína SMN procedente de SMN2 sólo se produzca completa en un 10 % de las casos.
En principio eso sería insuficiente para vivir a largo plazo, pero sucede que los seres humanos podemos tener desde cero hasta más de diez copias del gen SMN2.
Haciendo un cálculo rápido, si tenemos diez copias, y cada una produce un 10 % de SMN, generaremos el 100 % de la proteína SMN que necesitamos.
¿Pero qué pasa si tenemos menos copias de SMN2, pongamos cinco? Pues que se compensa sólo el 50 % del SMN necesario.
Así, en general, quienes carecen del gen SMN1 y tienen una sola copia de SMN2 padecen un caso grave de la enfermedad, con progresión más rápida (tipo I).
Quienes cuentan con dos copias sufren un caso más leve (tipo II).
Finalmente, con tres o más copias tenemos un caso aún más leve (tipo III) y se amplían las posibilidades de vivir más años.
Ante esta realidad podemos concluir que la evolución "se inventó" SMN2 en humanos (la copia de SMN1) para tener un salvavidas en el caso de perder el gen principal. Bonito, ¿verdad?

Tratamientos a precios desorbitados
Sabiendo todo lo que sabemos, ¿cómo podemos curar la AME? Si falta SMN1 o el gen está mutado, igual que pasa con otras enfermedades genéticas, no queda otra que sustituir la falta del gen.
De hecho, aunque suene a ciencia ficción, esta sustitución es algo que ya se hace. En 2019 se aprobó el medicamento Zolgensma, que permite introducir el gen SMN1 a través de un virus que lo lleva directamente hasta las motoneuronas.
Pero la auténtica belleza de la AME reside en que, gracias a que existe SMN2, podemos usar otras aproximaciones más originales.

Netflix lanza primer tráiler del live action de “One Piece” y confirma su fecha de estreno
Pensémoslo por un momento: si conseguimos averiguar qué hace que SMN2 sólo produzca un 10 % de proteína SMN completa y reparamos el problema para que el porcentaje sea mayor, podremos compensar la falta de SMN1.
Y eso es lo que hace el primer tratamiento que se aprobó en 2016 contra la AME, Nusinersen, y también otro más aprobado en 2020, Risdiplam.
Lo que consiguen estos tratamientos parece realmente fácil de hacer si nos apoyamos en un poco de biología básica.
En el caso de Zolgensma, se trata de llevar el gen SMN1 intacto hasta las motoneuronas, lo que actualmente se denomina terapia génica.
Y en el caso de los otros dos tratamientos citados, el reto es conseguir que algo se pegue a la región que diferencia a SMN1 y SMN2, para convertir al segundo en el primero.
Pero la dificultad de lograr que esto funcione, y además llevarlo al mercado de las enfermedades raras, hace que ambos tratamientos estén entre los más caros del mundo.
Zolgensma cuesta nada menos que… ¡dos millones de euros por dosis! Eso la convierte en la enfermedad más cara del mundo. Menos mal que sólo se precisa de una dosis.
En el caso de Nusinersen y Risdiplam, su coste oscilaría entre US$440.000 y US$876.000 por año (y, por supuesto, por paciente).
¿A qué se deben estos precios siderales? La respuesta fácil es que nunca lo sabremos. Las farmacéuticas suelen negociar el precio del medicamento de manera independiente en cada país y con transparencia cero.
Eso les lleva a considerar el medicamento como una mercancía, cuyo precio no tiene por qué estar ligado a su coste de desarrollo y producción.
Más bien es el precio que se está dispuesto a pagar por la vida y el bienestar de los pacientes en cada lugar del mundo.
Y se me olvidaba algo importante: ninguno de los tres medicamentos cura. Sólo hacen que la progresión de la enfermedad sea más lenta -lo que no es poco-.
Futuro para la AME y otras enfermedades raras
¿Qué va a pasar en el futuro con estos tratamientos frente a enfermedades raras?

Pues que, evidentemente, el mercado acabará haciendo que se abaraten los precios. Tratamientos como la terapia génica han sido usados ya para otras enfermedades, y sin duda seguirán apareciendo y, lo más importante, perfeccionándose.
Finalmente, la universalidad de su uso favorecerá la bajada de precios.
Por otro lado, podemos depositar nuestras esperanzas en el reposicionamiento de fármacos, es decir, el uso de fármacos ya existentes para otras enfermedades. Tiene la enorme ventaja de que abarata mucho los costes y permite acortar los tiempos de investigación.
En conclusión, es indiscutible que la AME es una enfermedad devastadora, y que los pacientes de AME siempre tendremos derecho a quejarnos de la mala suerte que tuvimos al nacer.
Pero, al menos, espero haber mostrado que esta enfermedad tiene un encanto especial a nivel molecular que, a quienes la padecemos, nos da también cierto derecho a tildarla como "la enfermedad más bonita del mundo".
*Antonio J. Pérez Pulido es profesor Titular de Universidad e Investigador en Bioinformática, Universidad Pablo de Olavide



